|
|
GENERALIDADES SOBRE LA
SORDERA NO SINDROMÁTICA
La pérdida auditiva es la alteración sensorial más común en humanos, se
estima que más de 70 millones de personas alrededor del mundo tienen una
pérdida que afecta la comunicación normal (1). La incidencia de hipoacusia
congénita severa es de al menos 1 en 1000 nacidos vivos (1,2), de estos
aproximadamente la mitad se atribuyen a factores genéticos, clasificándose
estos en, aproximadamente, un 70% como no sindromático y un 30 % como
sindromático (3). Para Colombia no se conocen datos epidemiológicos al
respecto, sin embargo en un país vecino, Brasil, un estudio reportó que la
hipoacusia no sindromática debido a mutación en un gen especifico,
Conexina 26, tiene un comportamiento similar al descrito en países
desarrollados (4). Se presentan estimaciones basadas en datos demográficos
de Colombia para el año 2002 (tabla 1). (5)
La
sordera no sindromática ocurre aisladamente, mientras que en
los casos sindromáticos se asocia con anormalidades en otros sistemas. Se
han descrito varios cientos de síndromes que cursan con hipoacusia y se
han encontrado las alteraciones genéticas en algunas formas frecuentes
(1-4,6).
La hipoacusia no sindromática se puede clasificar según su modo de
transmisión en: autosómica dominante, autosómica recesiva, ligado a X y
mitocondriales (1-3,6). Se presenta una clasificación según la etiología
de la hipoacusia y se muestran las frecuencias aproximadas según
diferentes fuentes
(tabla 2). (1-3) En los últimos años se han hecho importantes avances en
el diagnóstico de las hipoacusias no sindromáticas, se han encontrado los
locus genéticos, y sus productos proteícos, responsables de un importante
número casos. Al día de hoy se han identificado 51 locus para las formas
autosómicas dominantes, 17 de los cuáles se han clonado; 39 para las
autosómicas recesivas, con 17 genes clonados; 8 para las ligadas a X con
un gen clonado y 2 para las formas de transmisión mitocondrial que están
clonados (6). La nomenclatura internacional utilizada para denominar a los
locus genéticos de estas diferentes formas de hipoacusia designa DFNA (Deafness
A) a las formas de transmisión autosómica dominante, DFNB (Deafness B) a
las autosómicas recesivas y DFN (Deafness) aquellas con transmisión ligada
a X. Adicionalmente se coloca un número consecutivo, según el orden
cronológico de su descripción, por ejemplo los locus relacionados con la
forma de transmisión autosómica dominante se designan de DFNA1 hasta
DFNA51 (6).
FISIOLOGÍA AUDITIVA DE LA
SORDERA NO SINDROMÁTICA
El estudio genético de los pacientes con hipoacusia ha ayudado a aclarar y
ampliar el conocimiento de la fisiología del oído interno en su porción
coclear, incluso se han descrito procesos no sospechados hasta encontrar
su alteración(2,7,8). La cóclea es el sitio en donde la energía mecánica
de los ondas sonoras es convertida en potenciales de acción del nervio
coclear, iniciando así la transmisión de la información auditiva hacia los
centros del tronco cerebral y a centros superiores en la corteza cerebral,
proceso necesario para la comprensión e interpretación de los sonidos. La
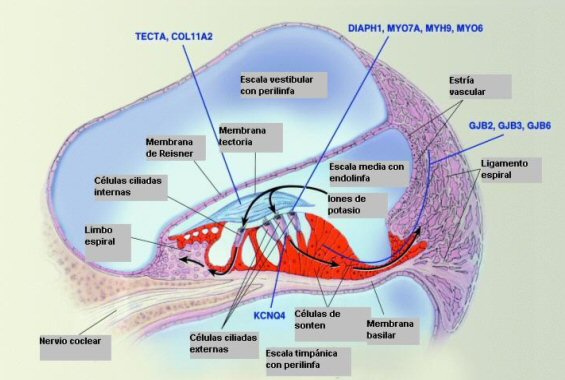
cóclea está localizada en la porción petrosa del hueso temporal, en su
longitud esta divida en tres compartimentos: la escala vestibular, la
escala media y la escala timpánica. La escala media, contiene endolinfa
con altas concentraciones de potasio y bajas concentraciones de sodio;
esta discrepancia en la composición electrolítica de los líquidos del oído
interno genera una diferencia de potencial eléctrico entre el interior y
el exterior de la célula, que juega un papel central en el proceso de
transducción de la información que se lleva a cabo en la cóclea. (Fig. 1).
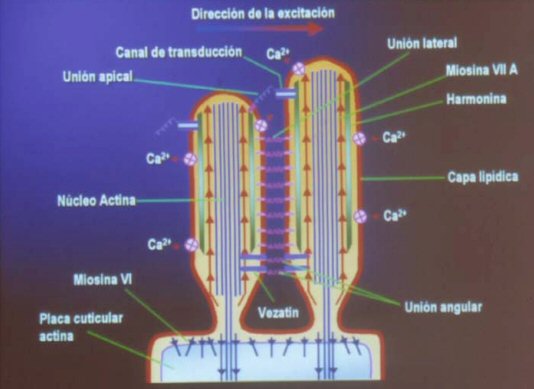
Los receptores auditivos son las células ciliadas del órgano de Corti. El
extremo de las estereocilias de las células ciliadas externas están
embebidas en la membrana tectoria, estas cilias tienen un esqueleto de
actina y formas no convencionales de miosina que están fijas a una lámina
cuticular rica en actina que a su vez sujeta la estereocilia al
citoesqueleto celular. Las estereocilias están ancladas unas a otras cerca
de su ápice de forma tal que se mueven en conjunto. Las células ciliadas
internas son los receptores primarios y reciben la mayoría de las fibras
aferentes del nervio coclear; las células ciliadas externas, reciben la
mayor parte de la información eferente del mismo nervio, tienen por
función promover la discriminación de frecuencia y amplificación de la
señal, de forma que modulan el funcionamiento del receptor primario
(2,7,8). (Fig. 2 y 3)
Los movimientos de la membrana timpánica, en respuesta a las ondas
sonoras, son transmitidos y amplificados por la cadena oscicular y
retransmitidos como ondas de compresión hacia la escala vestibular de la
cóclea. Estas ondas mueven la membrana basilar causando la deflexión de
las estereocilias contra la membrana tectoria. La deflexión de las
estereocilias conducen a la apertura de canales iónicos que permiten la
entrada de potasio al interior de la célula ciliada induciendo su
despolarización. La despolarización celular genera la activación de
canales de calcio conllevando a la movilización de vesículas sinápticas y
posterior liberación del neurotransmisor en el espacio sináptico; de esta
forma se inicia la activación del nervio coclear.
Las moléculas de miosina no convencional juegan un papel importante en el
proceso de transducción manteniendo la tensión entre las uniones de los
ápices de las estereocilias.
Para mantener el funcionamiento de la célula ciliada, los iones de potasio
que entran en su interior debe salir, además debe mantenerse una alta
concentración a nivel de la endolinfa. Con el fin de mantener este proceso
se ha descrito un mecanismo de reciclaje del potasio mediante el cual
estos iones salen de la célula ciliada a nivel de su membrana basolateral
por un canal de potasio, alcanzando las células de soporte del órgano de
Corti. Posteriormente difunden en forma pasiva de célula a célula a través
de uniones brecha (gap junctions) compuestas por una proteína multimérica,
denominada conexina 26, presente en las células de soporte del órgano de
Corti, células del limbo y del ligamento espiral. Una vez los iones de
potasio alcanzan la estría vascular son activamente bombeados hacia la
endolinfa por canales de potasio dependientes de voltaje. (2,7,8)
La membrana tectoria es una estructura acelular con una función mecánica
en el proceso de transducción de la señal, conformada por una matriz
proteíca. Varios tipos de colágeno forman más de la mitad de su
estructura, predominando el colágeno tipo II, y menores cantidades de tipo
IX y XI. La proteína no colágeno más abundante es la α tectorina.
FISIOPATOLOGÍA DE LA
SORDERA NO SINDROMÁTICA
Existen 5 sitios en donde con mayor frecuencia se producen alteraciones
funcionales y estructurales del órgano de Corti que conllevan a un
malfuncionamiento bioquímico del mecanismo de audición (Tabla 3). Estos
sitios son:
1.Alteraciones de los componentes de la membrana y proteínas importantes
en el equilibrio endolinfático.
Varias moléculas se han identificado en el mantenimiento del equilibrio
iónico endolinfático. Una de las más importantes es la Conexina 26 (Cx26),
no solo porque su mutación fue una de las primeras descritas en casos de
sordera no sindromática sino también porque representa la mayor causa de
este tipo de alteraciones entre diferentes poblaciones estudiadas (DFNB1),
representando en algunos casos mas del 50% de las sorderas no
sindromáticas de transmisión recesiva (2,7-9). El gen que la codifica se
ha designado GJB2. El fenotipo mas comúnmente asociado es una hipoacusia
neurosensorial prelingual severa a profunda, con variabilidad intra e
interfamiliar. Mutaciones en la conexina 26 también se han descrito en
casos sordera no sindromática autosómica dominante (DFNA3) y de sordera
sindromática (síndrome de Vohwinkel: queratodermia y sordera).
La conexina 26 es una molécula estructural presente en la membrana
basolateral que forma las uniones brecha. El ensamblaje de seis
subunidades de conexina forman una estructura llamada conexón, el empalme
de dos conexones adyacentes establecen una unión brecha a través de la
cual células contiguas intercambian moléculas de pequeño tamaño como
iones. Estas uniones a nivel de la cóclea se ha encontrado en la estría
vascular, membrana basilar, limbo y ligamento espiral. La Cx26 juega un
papel central en el mecanismo de reciclaje del potasio. La mutación más
común es la deleción de guanina en la posición 35, (35delG), también
llamada 30delG, esta mutación se ha encontrado en más de dos tercios de
las personas con DFNB1 en poblaciones de Italia, Israel, Pakistán, España,
Francia, India, Caucásicos, y en Árabes (1-4,7-10). Se ha reportado que
mutaciones en la Cx 26 pueden ser un factor agravante en la toxicidad por
aminoglucósidos en pacientes con sordera no sindromática de transmisión
mitocondrial (11). Mutaciones en otras conexinas, Cx30 (DFNB1), Cx31
(DFNA2), Cx 43 han sido descritas en algunas familias, tanto en herencia
recesiva como dominante (9, 12).
Otros genes involucrados en el reciclaje del potasio pero cuyo mecanismo
de transmisión es autosómico dominante es el KCNQ4 (DFNA2), que codifica
un canal de potasio importante en la remoción de este ión de las células
ciliadas (2,6,7). El gen KCNQ1 (o KCNE1) codifica para un canal de potasio
importante en la secreción de este ión hacia la endolinfa. Su mutación se
asocia con el síndrome de Jervell y Lange-Nielsen (defecto cardiaco y
sordera) (2,6).
Mutaciones en el gen PDS se encuentra tanto en casos de sordera no
sindromática (DFNB4), como en el síndrome de Pendred (sordera y
alteraciones tiroideas), el cual es la causa más común de sordera
prelingual sindromática. Su producto, la pendrina, es un transportador de
cloro y yodo independiente de sodio que se expresa tanto en el oído
interno como en la glándula tiroides; su mutación en animales de
experimentación produce dilatación del compartimiento endolinfático y
defecto otoconial, lo cual supone un rol en la homeostasis iónica del oído
interno (6-8).
Una proteína denominada Claudin-14 se encuentra mutada en casos de DFNB29.
Esta proteína forma uniones estrechas intercelulares, importante mecanismo
de barrera y modulador de la permeabilidad transcelular. Actúa como lÍmite
entre las membranas apical y basolateral, manteniendo los gradientes
electrolíticos y diferencia de potencial entre la endolinfa y las células
del órgano de Corti, para permitir la despolarización de las células
ciliadas. (6-8)
2. Alteraciones moléculares del citoesqueleto celular En este grupo
encontramos tres genes que codifican un tipo de miosinas llamadas no
convencionales porque difieren de las encontradas en las células
musculares. Estas son: la MYO7A, MYO15 y MYH9; sus mutaciones se asocian
con DFNB2, DFNB3 y DFNA11. En el oído interno las miosinas no
convencionales se encuentran en las estereocilias y en la lámina cuticular
de las células ciliadas; junto con la actina juegan un papel importante en
la organización de la estereocilia y en el movimiento de las uniones de
los extremos de las estereocilias, estructura crucial en el flujo de
cationes durante la transducción de la señal.
Las mutaciones en MYO7A se han identificado en el síndrome de Usher tipo
IB (sordera congénita, disfunción vestibular y retinitis pigmentosa) (1,
6-8).
Mutaciones en el gen Diaphanous (DIAPH1) se ha identificado en pacientes
con DFNA1. Su producto genético pertenece a la familia de las forminas
involucradas en la citocinesis y el establecimiento de la polaridad
celular, se cree que regulan la polimerización de actina y ayudan a
mantener el citoesqueleto de ésta en las células ciliadas (2,6-8).
3.Alteraciones de moléculas estructurales del órgano de Corti y de la
matriz extracelular. Las proteínas de la familia del colágeno son
moléculas heterogéneas codificadas por más de 30 genes diferentes. A nivel
del órgano de Corti la mutación en el gen para una de ellas, el COL11A2,
se asocia con DFNA13 y una forma de síndrome de Stickler (malformaciones
faciales, alteraciones oculares, artritis e hipoacusia). Este gen codifica
para la subunidad α2 del colágeno 11, molécula importante para mantener la
integridad estructural de la membrana tectoria. Fenotípicamente se
presenta como una sordera no sindromática no progresiva que afecta las
frecuencias medias (1,2, 6-8).
La α-Tectorina es una molécula que interactúa con β-Tectorina para formar
parte de la matriz no-colágena de la membrana tectoria. Mutaciones en su
gen, TECTA, se asocian con varios tipos de sordera no sindromática: DFNA8,
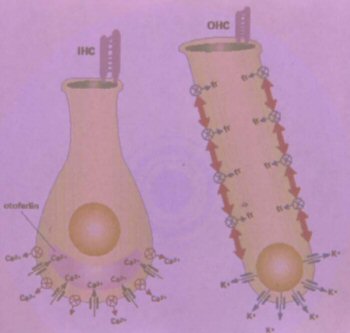
DFNA12 y DFNB21 (2, 6-8). OTOF es un gen que codifica para un producto
llamado otoferlina, proteína citosólica anclada a la membrana de la base
de las células ciliadas internas, en la región sináptica. Se cree que está
involucrada en el tráfico de vesículas sinápticas. Mutaciones en este gen
se han encontrado en pacientes con DFNB9 (6).
El Gen COCH codifica para un producto que parece ser una proteína
extracelular encontrada en el ligamento espiral y en el estroma del
epitelio vestibular, se cree que es importante en el mantenimiento de las
otras proteínas estructurales de la cóclea. Su mutación causa una forma de
sordera sindromática dominante, DFNA9, progresiva, de establecimiento
tardío y asociada a compromiso vestibular; pueden presentarse cuadros
similares a la enfermedad de Meniere, incluyendo vértigo, tinnitus y
plenitud aural, hasta en un 25% de los pacientes (2,6-8).
4. Alteraciones en proteínas involucradas en otros procesos celulares El
gen POU4F3 codifica para un miembro de la familia de los factores de
trascripción, importantes en el proceso de regulación de la expresión de
otros genes; este producto genético es requerido para la maduración,
mantenimiento y supervivencia de las células ciliadas. Su mutación conduce
a un tipo de sordera progresiva autosómica dominante de establecimiento
tardío, DFNA15 (1,6).
Otro regulador del desarrollo celular, el producto del gen POU3F4, se ha
encontrado mutado en familias con sordera congénita mixta, conductiva y
neurosensorial; su mecanismo de transmisión es ligado a X, DFN3, y se
encuentra en pacientes que presentan fijación estapedial y una anormal
comunicación entre el liquido cefalorraquídeo y la perilinfa (6).
5. Alteraciones en los genes mitocondriales En contraste con el genoma
nuclear, el genoma mitocondrial contiene solo información para codificar
13 proteínas, 22 tRNAs (RNA de transferencia), y 2 rRNAs (RNA ribosomal).
Las mutaciones en su genoma se caracterizan por un patrón de herencia
materna. Respecto a la hipoacusia congénita se ha visto tanto casos
sindromáticos como no sindromáticos. En Los cuadros sindromáticos se
asocian a sordera congénita con episodios de encefalopatía, acidosis
láctica, miopatía, diabetes mellitus, oftalmoplejía, ataxia y atrofia
óptica. La mutación en el gen de 12S rRNA y tRNAser pueden conducir a
sordera no sindromática. Así mismo, la mutación en el gen 12 S rRNA,
también se asocia con susceptibilidad a los aminoglucósidos, conduciendo a
hipoacusia en aplicaciones de dosis que normalmente no afectarían la
audición (1, 2, 6-8).
CONCLUSIONES
La aproximación genética al estudio de la sordera ha probado ser una
poderosa herramienta en la comprensión de las bases moleculares de la
función auditiva.
Procesos considerados vitales en el funcionamiento de la cóclea como el
mecanismo de reciclaje del potasio se han dilucidado gracias al estudio de
alteraciones en los productos de los genes que los regulan. Un aspecto
quizás más importante desde el punto de vista práctico es la utilización
de este conocimiento en la consejería genética, detección y tratamiento
temprano de pacientes, y finalmente en el establecimiento de la terapia
genética (1, 13).
Hay aspectos importantes que ayudan a cumplir estas metas en el caso de la
sordera no sindromática, como por ejemplo, estudios en diferentes
poblaciones han mostrado que la mutación en una sola proteína, Cx26, es
responsable de la mayoría de casos de sordera no sindromática recesiva;
además una mutación particular, 35delG, representa alrededor del 70% de
todas las mutaciones.
Finalmente la Cx26 está codificada por un solo exón, lo cual facilita el
tamizaje. No hay duda que el diagnóstico genético es una herramienta
a la que se debe acudir en los casos de sordera congénita de origen
genético, y probablemente la detección de mutaciones en el gen de la Cx26
sea un primer paso razonable cuando nos encontremos ante pacientes con
sordera no sindromática de transmisión recesiva.
Ante este panorama es indudable que se hace necesario realizar estudios
tendientes a aclarar la situación de nuestro país con respecto a las
causas de sordera congénita de origen genético. |
|